Will your PR ever be Merged?
P
Staff Engineer at Instruqt
Search for a command to run...
Staff Engineer at Instruqt
I did a quick check on Vue.js and it looks like the figures are caused by spam PR's , see https://blog.domenic.me/hacktoberfest/
So your figures say something about the percentage of PR's being merged, but that does not have to say anything about the hospitality of the project ;-)
You are right. I am planning to exclude PRs with label spam and bot made PRs. Thanks for looking into this!
I made a few issues on merge-chance GitHub repo, including for the problem you mentioned: https://github.com/PiotrZakrzewski/merge-chance/issues Feel free to give feedback / your ideas!
Piotr Zakrzewski You might want to mention near the start of the current version of this article that you are working on refining your method because of your new insights. Especially the conclusion should be reworded in my view ;-)
done, again thanks for your feedback. Hans Klunder
Hans Klunder Have a look at this: https://acceptance.merge-chance.info/target?repo=vuejs/vue (still not officially released version) even after accounting for spam (mostly ..) the merge chance for the old Vue version is really low .. Now you can download the data that it takes into account to verify (still a bit of spam but no longer dominant).
Piotr Zakrzewski (disclaimer: I am in no way related to the Vue project, just took this one as an example ;-)) I downloaded your data on Vue and found some details:
1) you only have PR's from 2020 whereas Vue's PR's start at 2014 2) it is indeed the old version 2.x version of Vue
Popular projects can attract way more submitters, but that does not mean those are all good developers e.g.: 1) some PR's are closed because of submitter tries to fix something that is not broken (e.g. see https://github.com/vuejs/vue/pull/11595 ) 2) some PR's are closed because the submitter does not provide info to reproduce the error that the PR would fix (e.g. https://github.com/vuejs/vue/pull/11598)
So while the numbers might be correct, it does not automatically prove that a project is less hospitable. There might be other reasons for these figures. Correlation and Causation etc ;-)
Ps. if you still want to name Vue then using the current 3.x Repo might be more current https://acceptance.merge-chance.info/target?repo=vuejs/vue-next
Btw: there also seems to be something wrong with the calculations, e.g.:
https://acceptance.merge-chance.info/target?repo=JuliaLang/julia
It says your chance on getting merged is 91.67% However downloading the data gives me 60 records of which 57 were by contributors and got merged. 3 were by outsiders ("none" in your data) and did not get merged.
Based on this data I could say that JuliaLang/julia is rather inhospitable as JuliaLang/julia always merges from contributors but never from outsiders ;-)
Your chance on getting merged is (based on this data ;-)) 0% and not 91.67%
That 0% sounds rather odd to me (and so does 91.67% ;-)) so I would never publish strong conclusions without more research
Hans Klunder The CONTRIBUTOR authorAffiliation means only the someone merged anything before to the repo. Not that they are an insider. Only members and owners are currently excluded, plus those who merged more than 5 times recently.
None is (in most cases) a first time contributor. The Julia repo calculation seems alright to me :)
Hans Klunder What I want to achieve with my stats crunching (and merge-chance.info) is to help people find repos that are likely to accept their contributions. It is true that these (simple after all) stats won't take into account many cases, such as the one you mentioned - a contributor not knowledgeable enough - but I think it may still help tell an active and open project from a non-responsive and insider-focused one. Again, thanks for thinking along!
Piotr Zakrzewski the 5 merges rule is only on acceptance environment. The merge-chance.info version still runs the simple way of calculating stats.
Piotr Zakrzewski Thanks for clarifying this, that does indeed explain the 91.67%
The 5 merges rule sounds reasonable but without supporting evidence it could skew your results.
Once you got your first PR through then you know (and appearantly aligned with ;-)) the "culture" of the project (coding style, doc style etc, way of commenting). The project also gets to know you a bit ("that person from last PR") so you start to build trust. Both items will make it easier to get a second PR merged.
On the other hand: newbies with more ambition than skill will get their PR rejected and will never try again. Only the newbies with good enough skills and/or willingness to invest time will get their PR merged.
I have submitted to repo's where people asked me to rebase twice because they put their own PR's first. I got my PR merged, but it was not really a hospitable experience. Looking at the cold hard figures I got my PR merged, so it counts towards an increased chance of getting merged.
So although the figures are right (even the ones on Julia ;-)) I would not draw any conclusions on hospitality based on this data.
If you really want to know what your chances are on a succesful PR then my advice would be to get to know the project and raise an issue stating that you intent to submit a PR to do XYZ. If that issue goes stale or your intent is dismissed then you know enough ;-)
Personally I also prefer people to first submit an issue and discuss instead of starting stealth and suddenly submerge with a fully loaded PR that changes 75% of my project ;-)
And then some suggestions for other stats that could be useful:
Kind regards, Hans
Hi! FYI, you can obtain this data for all existing repositories by one request in one second: https://gh-api.clickhouse.tech/play?user=play#U0VMRUNUCiAgICByZXBvX25hbWUsIAogICAgdW5pcShhY3Rvcl9sb2dpbiksIAogICAgdW5pcShudW1iZXIpLCAKICAgIHN1bShtZXJnZWQpLCAKICAgIHJvdW5kKHN1bShtZXJnZWQpIC8gdW5pcShudW1iZXIpICogMTAwKSBBUyBwZXJjZW50IApGUk9NIGdpdGh1Yl9ldmVudHMgCldIRVJFIGNyZWF0ZWRfYXQgPj0gdG9kYXkoKSAtIElOVEVSVkFMIDkwIERBWSAKICAgIEFORCBldmVudF90eXBlID0gJ1B1bGxSZXF1ZXN0RXZlbnQnIApHUk9VUCBCWSByZXBvX25hbWUgCk9SREVSIEJZIHVuaXEoYWN0b3JfbG9naW4pIERFU0M=
Have you ever tried expressing board game rules in code? Does it sound a bit tedious? I bet it does, I implemented a few board game rulesets in Python and JavaScript to write a simulator or implement a version of the board game playable in a browser....
A painfully basic static code analysis example

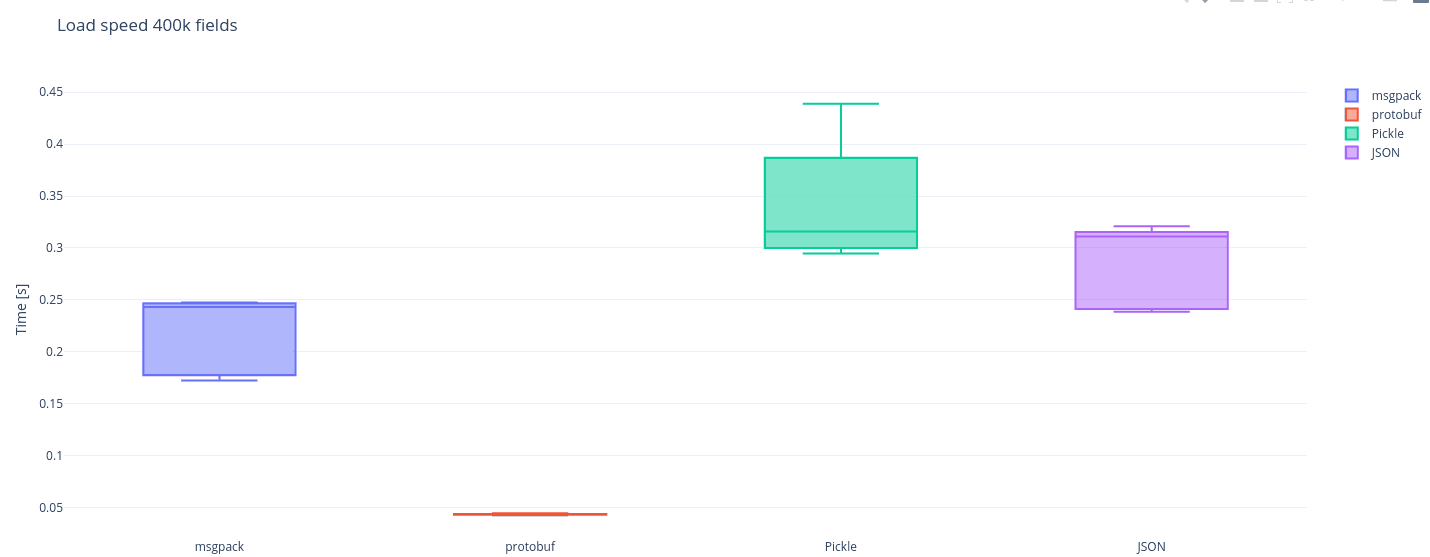
Disclaimer: This is a fairly specific benchmark, it addresses only serialization performance of a single big Python object. That was the problem we were solving at the time at Plotwise. It might not be relevant to any other case, but you can probably...
Harder than it sounds!
Learning big legacy codebases is a chore. I wish there were more tools to help me learn what is going on in an existing application. There is a lot of focus on designing and building new software, but when it comes to modifying existing ones, especia...
Some of the results (e.g. Vuejs) are skewed by spam. I am currently refining my methodology - if you are curious have a look at merge-chance GitHub issues
Have you ever made an open source contribution? Whether your PR is rejected/ignored or successfully merged can depend on factors other than just the quality of your work. Some projects are just much more responsive, some are very picky about what is accepted and reject anything that does not match their vision.
I extracted Pull Request stats for 40 popular open source GitHub projects to see how likely it is for a PR to ever be merged. In this post you will find contributions to which projects are the best use of your time. Spoiler: some big mature projects do better in this ranking than you would think!
I went with Python, Julia, R and JavaScript, each having 10 repos in the ranking. The aim was to collect a somewhat representative sample of big popular projects among these 4 languages, hence you will see here mostly big names such as React, TensorFlow and Shiny. I am personally interested in broadly understood Data Science so my ranking skews more to this side, since these are the projects I would be more likely to contribute myself. However an open source ranking would not be complete without popular web frameworks in Python (Django, Flask, FastAPI) and some of the most popular js frontend tech (React, Vue), since web development projects are the most active ones on GitHub.
To answer the question: how likely is it that an average PR gets merged into given repo I looked at proportion of Merged vs closed without merge PRs. I also collected data about stale PRs (open for longer than 90 days) and currently active ones (open but not older than 90 days).
This is an interesting one! At first I used GitHub's REST API and while it was good enough for extracting data from smaller repositories I quickly hit rate-limiting (5k requests per hour) and also got bored of waiting. Fortunately GitHub offers also a GraphQL API which for this specific purpose is orders of magnitude more efficient!
Why is that? For my analysis I need just a few fields from all PRs in a given repo. I used PyGithub lib to fetch the data, fetching PRs seems to incur one HTPP request per paginated result (max 25 entries) which is what I expected but then for fetching the merged status field it had to perform one more HTTP request per PR. As you can imagine that slowed down the execution to a crawl so I had to find another solution, GraphQL performed just one HTTP request per 100 entires and dropped execution time from above an hour for a big repo like react to just around a minute. Have a look at my extraction scripts (both GraphQL and REST) on my GitHub.
OK enough about the data collection. You are probably just curious which projects are most likely to ghost you and your hard work. Let get down to it.
First have a look at the list of JS projects I analysed:
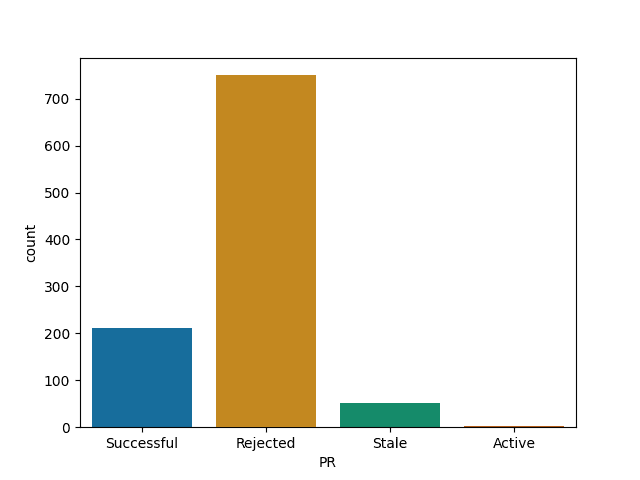
For each of them I report the number of successful (merged) PRs, rejected (closed but not merged), stale (open for longer than 90 days) and active (open and less than 90 days old).
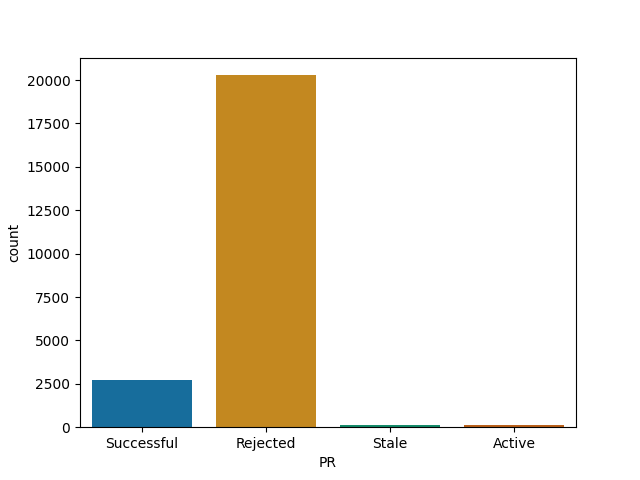
Express and Node are most likely to reject a PR
Express
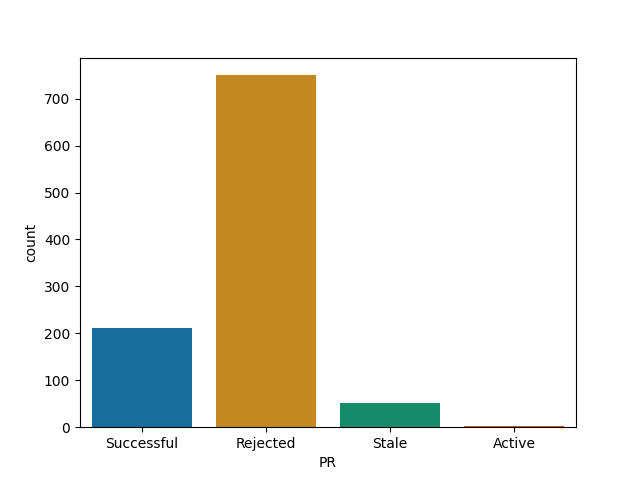 Node
Node
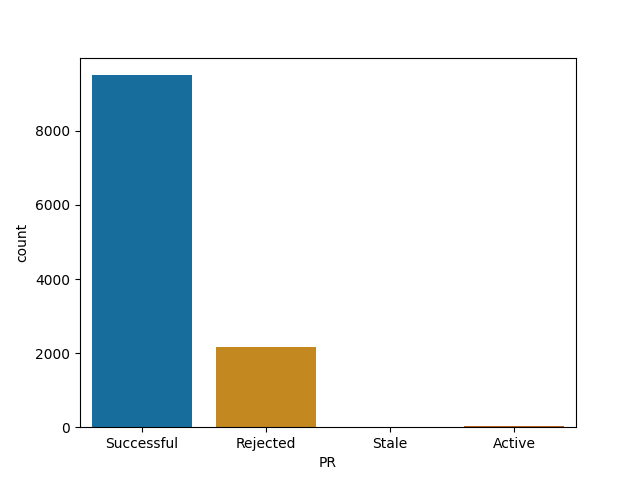
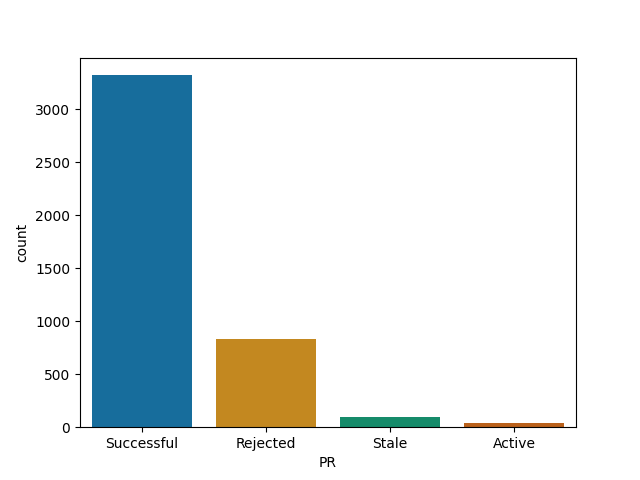
Material-ui and webpack are most likely to merge your PR among big JS projects
Material-ui
Webpack
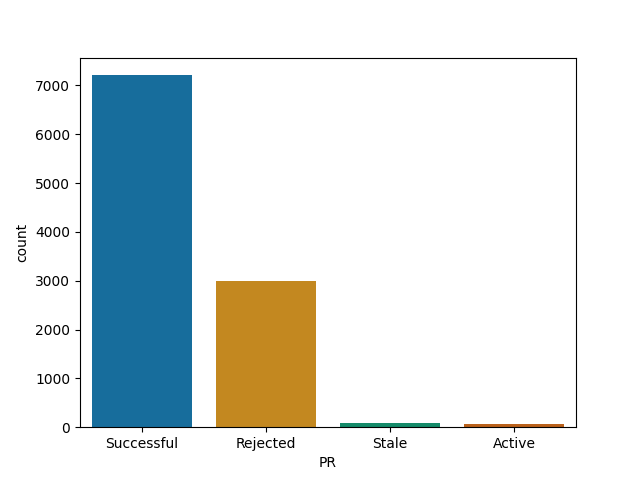
React, while quite selective is more likely to merge than I thought such a big project would be
React
Lets have a look at the list of Python projects I analysed:
For each of them I report the number of successful (merged) PRs, rejected (closed but not merged), stale (open for longer than 90 days) and active (open and less than 90 days old).
Big old web projects like Django and Flask are significantly more selective, the chance for merging is still higher than in JS equivalents
Django
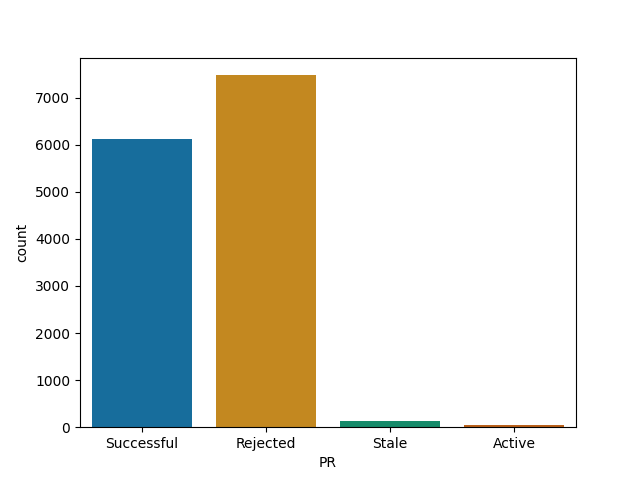
Flask
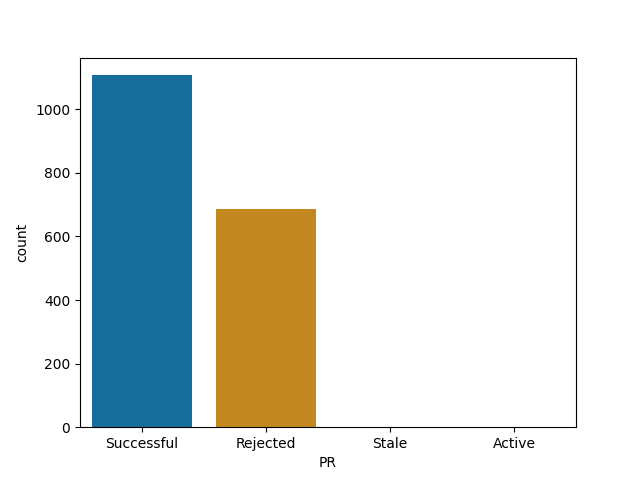
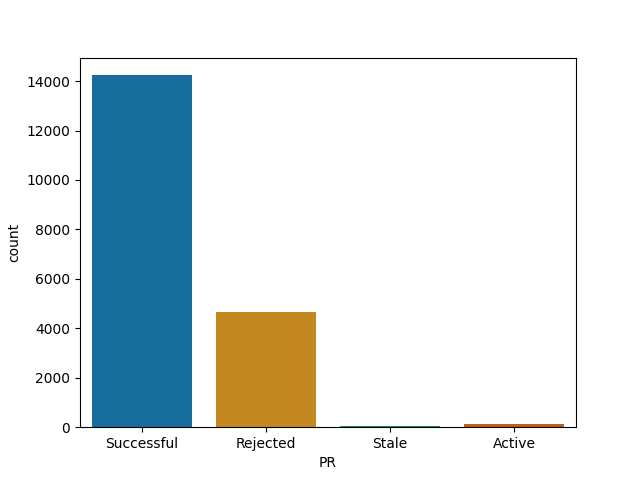
Data related projects merge most of their PRs
Pandas
Scikit-learn
DS Notebooks
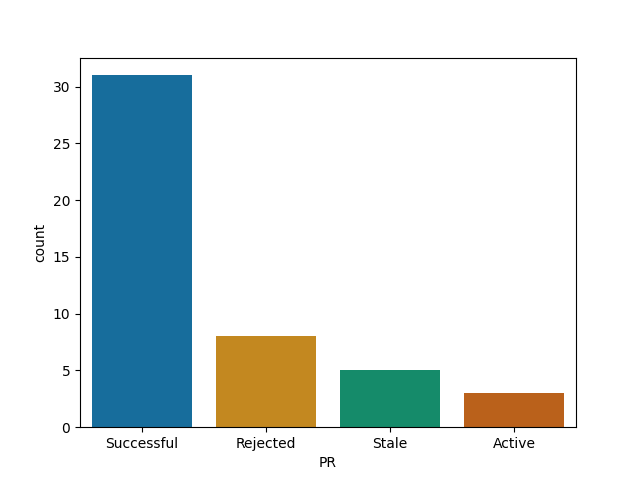
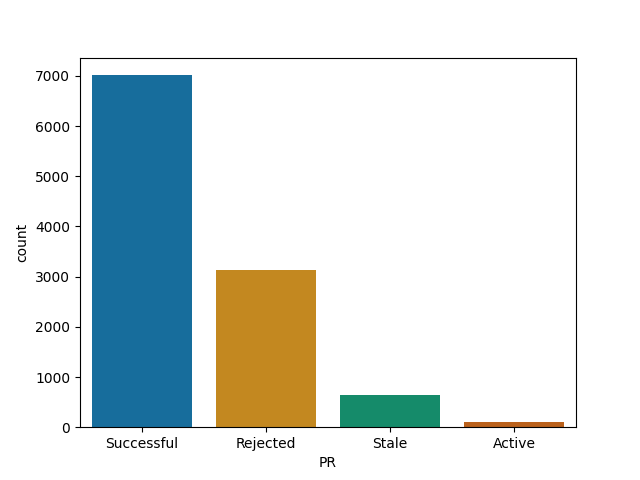
Tensorflow is more welcoming to contributions than I assumed ..
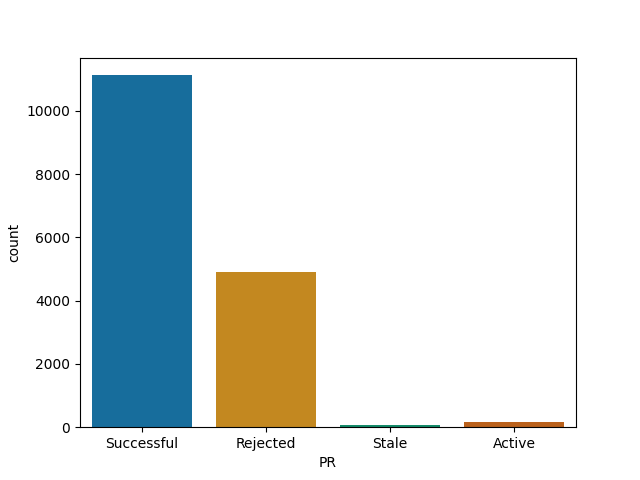
3blue1brown and his popular manim visualisation lib stand out
Manim was open sourced by popular math Youtuber 3blue1brown not so long ago. It seems that there aren't enough maintainers to process deluge of PRs coming from the community. Some help here maybe?
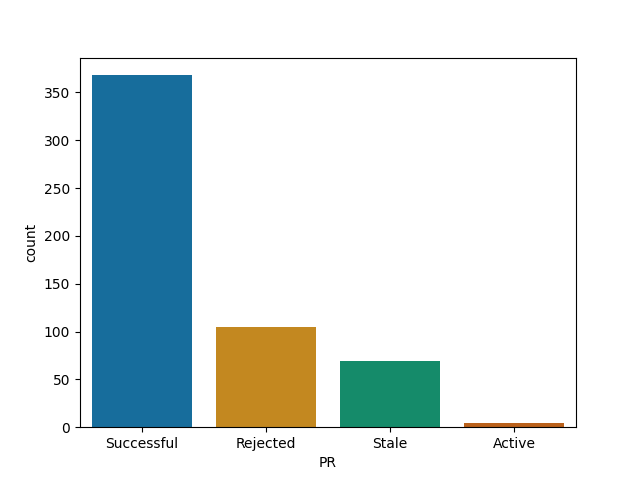
Lets have a look at the list of Julia projects I analysed:
For each of them I report the number of successful (merged) PRs, rejected (closed but not merged), stale (open for longer than 90 days) and active (open and less than 90 days old).
Roughly all Julia's repos follow similar pattern - your time is probably well spent here!
JuMP
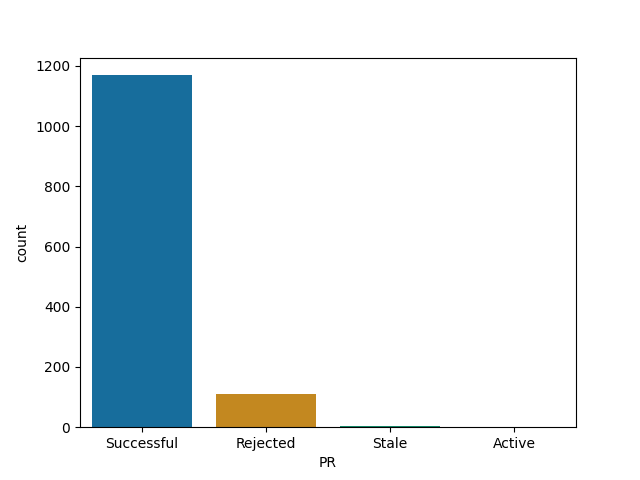
Pluto
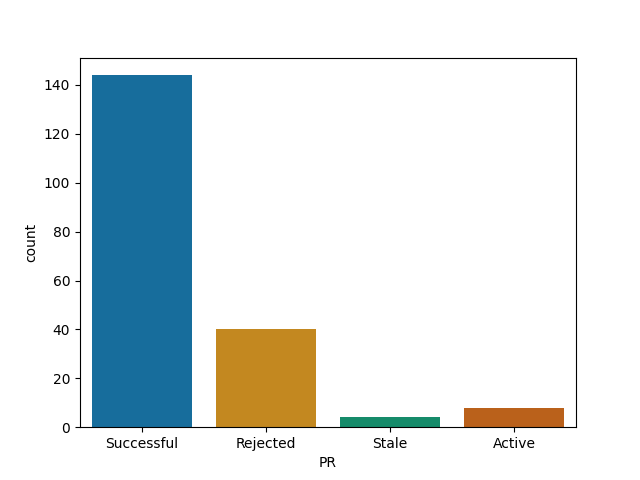
DifferentialEquations
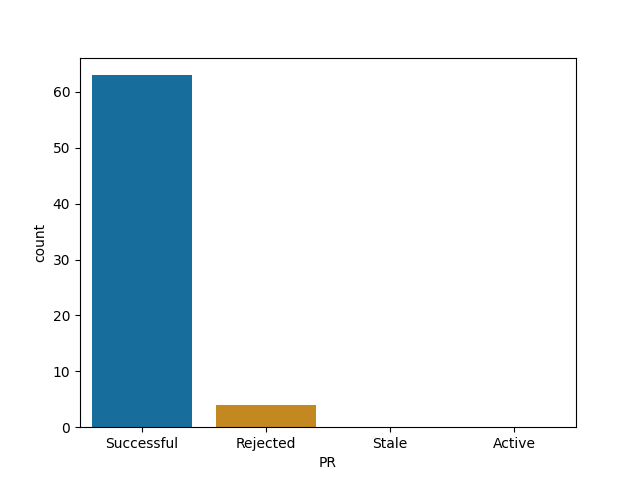
PyCall
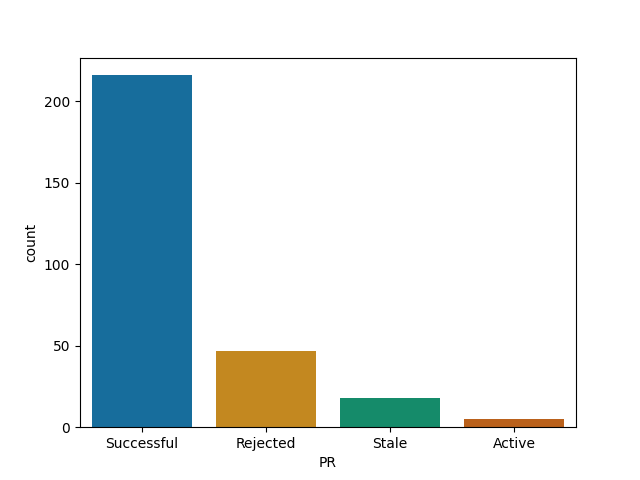
Plots.jl
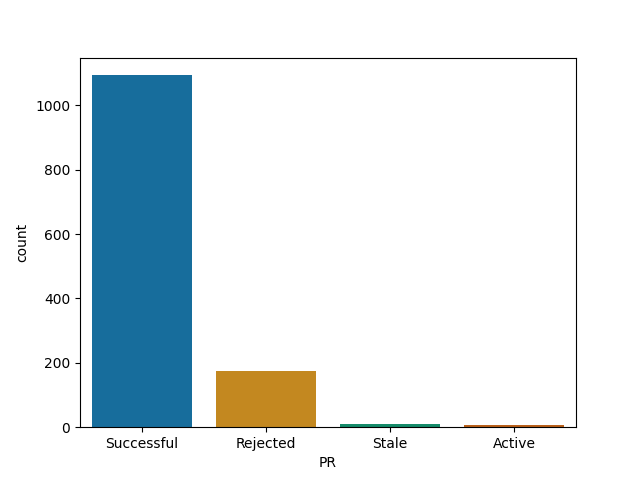
JuliaLang
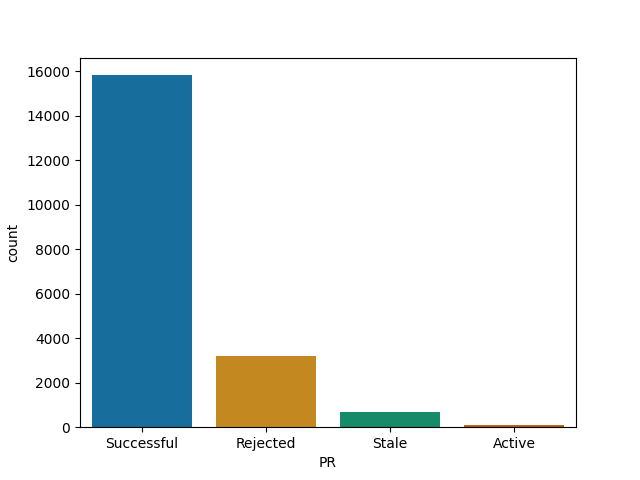
IJulia
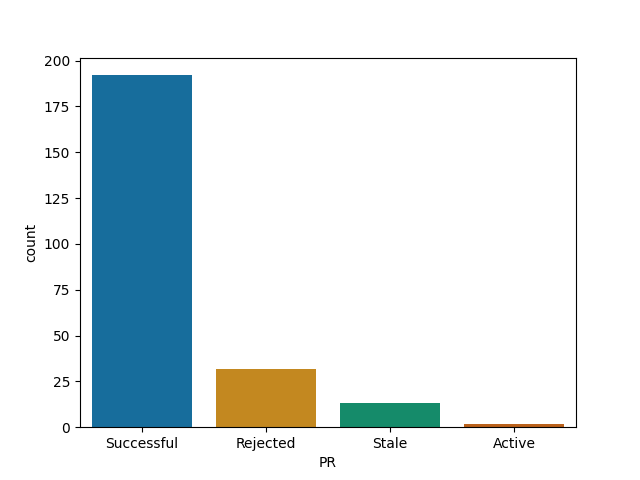
DataFrames
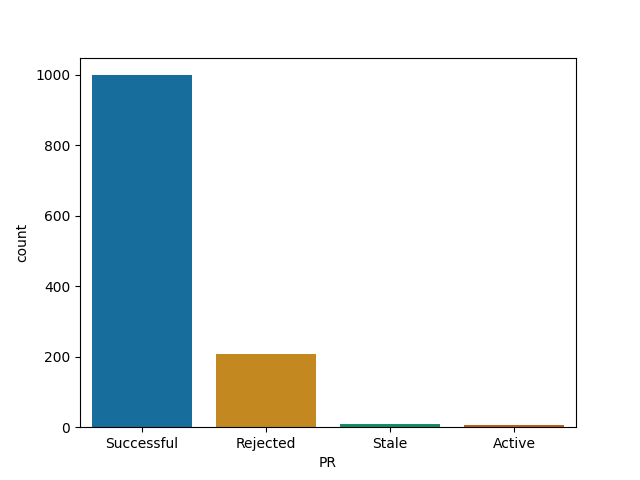
JuliaTutorials
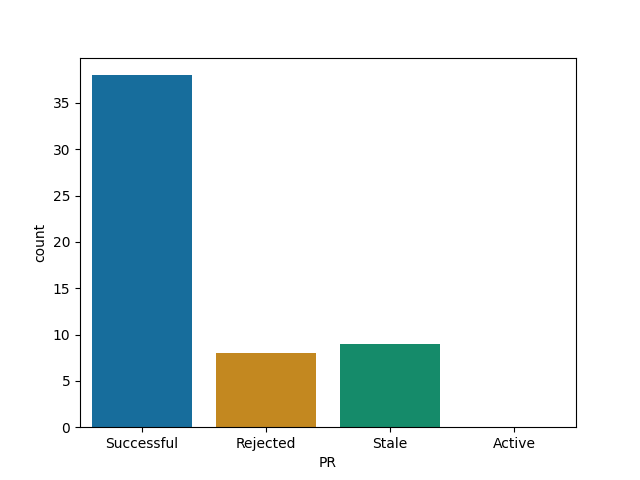
Gadfly
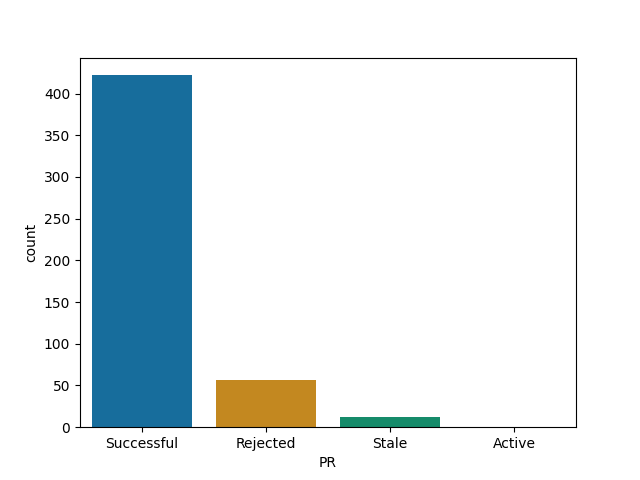
R projects I analysed:
For each of them I report the number of successful (merged) PRs, rejected (closed but not merged), stale (open for longer than 90 days) and active (open and less than 90 days old).
Knitr
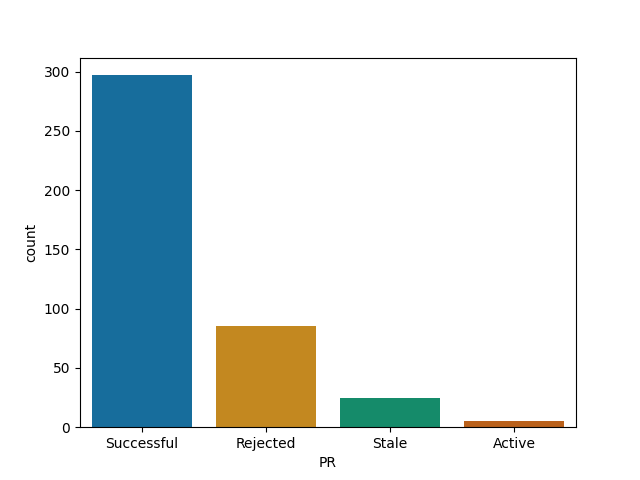
ggplot2
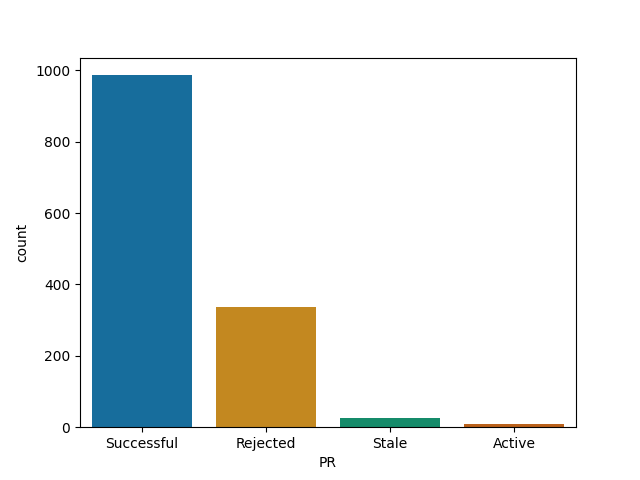
dplyr
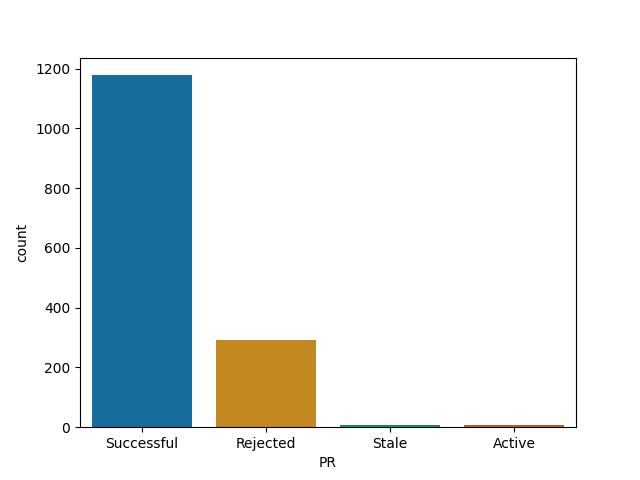
shiny
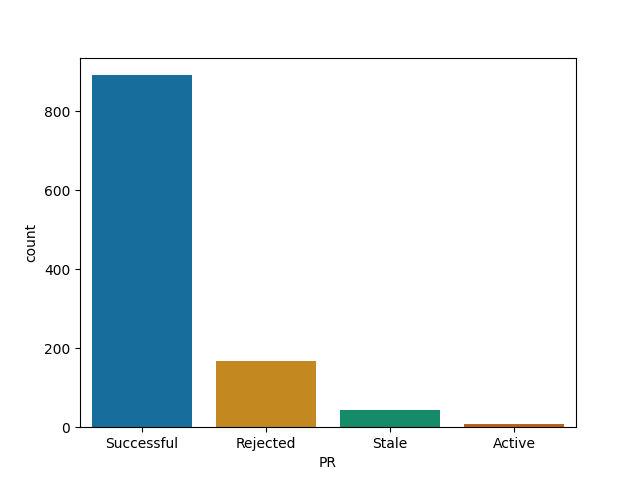
rmarkdown
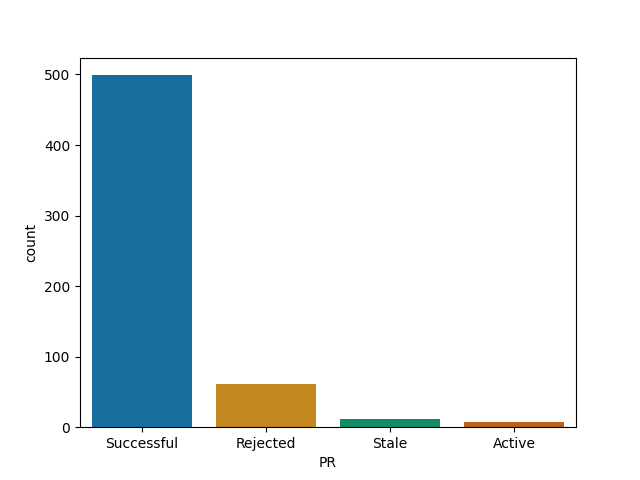
plotly
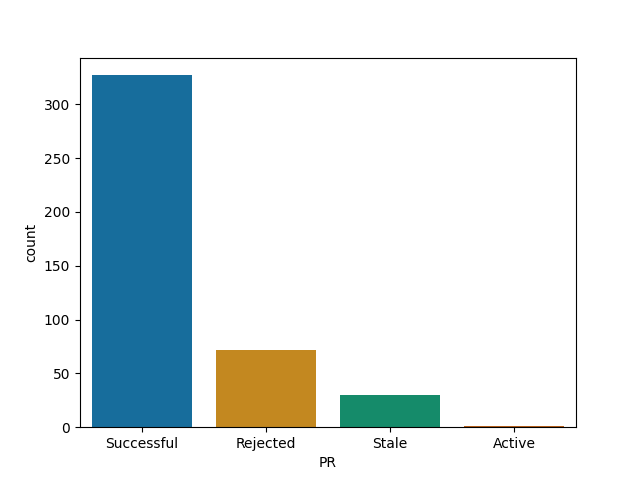
DiagrammeR
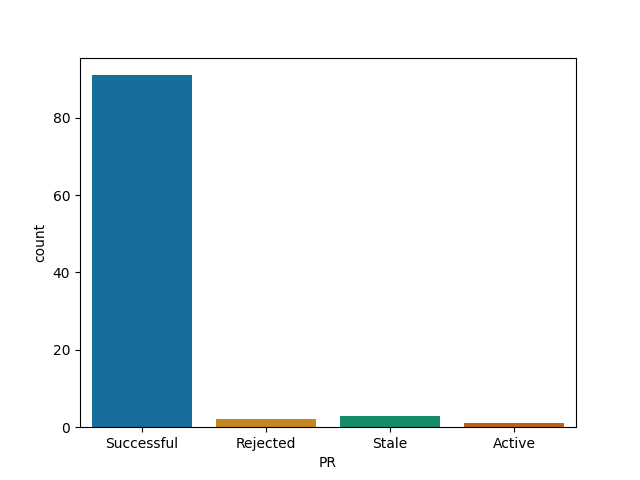
devtools
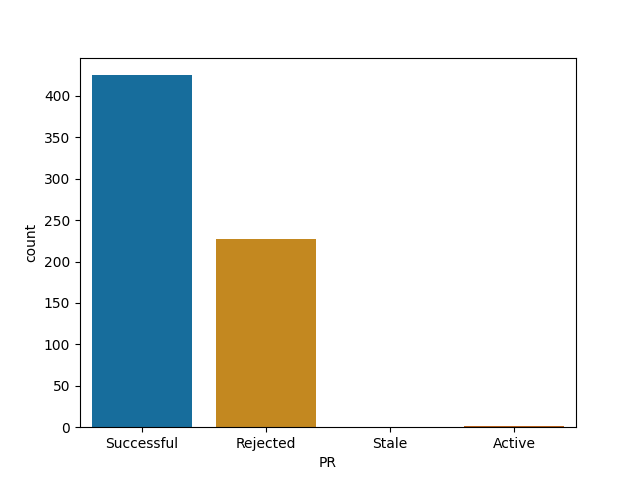
MLR
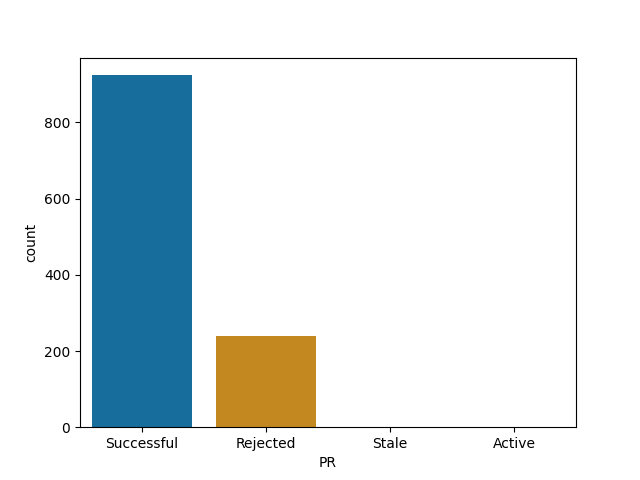
r4ds
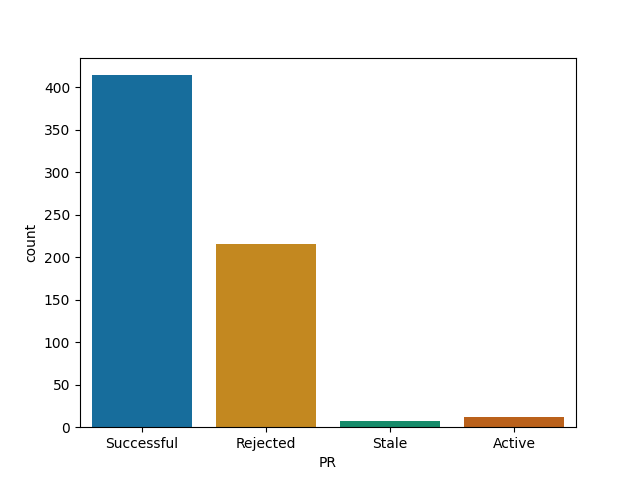
Based on this limited analysis (it is in the end just a ratio of merged vs other PRs) it seems that big JavaScript projects are less good of an investment of your time when it comes to making a PR, although the analysis does not take spam into account yet, so take it with a grain of salt. Julia with its rapidly developing ecosystem is quite an attractive target for contributions however!
Would you like to get a PR stats plot like above for some other repo? Use my script.
Follow me on twitter for more content like this!